")
FDA’s 505(b)(2) Application
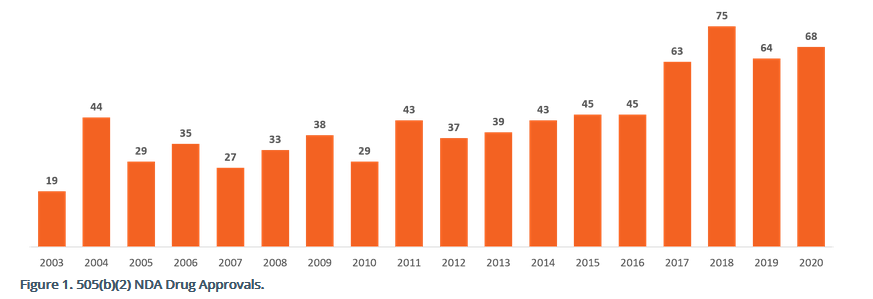
The 505(b)(2) new drug application (NDA) is an attractive regulatory and commercial strategy for clients. This hybrid NDA application helps avoid duplicative and costly studies that others have already completed. FDA’s rules allow (b)(2) applicants to rely on this prior body of work. And because of this reliance, the (b)(2) development is cheaper and faster towards full FDA approval when compared to the traditional (b)(1) route. The following bar graph shows the FDA’s approvals of 505(b)(2) applications.
Benefits of the 505(b)(2)
Most people think that drug products are either completely brand new drugs consisting of brand new molecules or generic drugs (essentially copies of branded drugs). Sometimes, however, a new drug is neither strictly a generic, nor an entirely new molecular entity. In such cases, the 505(b)(2) application may be the pathway of choice. If the drug is similar to an existing drug but has certain new traits, such as a new form of dosage, strength, indication, or how it interacts with other drugs, the 505(b)(2) pathway is likely a superior option to the typical NDA under section (b)(1).
Drug manufacturers can submit their application to the FDA using data collected by another manufacturer, but it is up to the submitting manufacturer to show a connection between the reference drug and their own. If this connection can be shown, the FDA may approve the new drug without requiring new pre-clinical studies or major clinical trials. For example, if a branded drug is already on the market in immediate release form, then it makes no sense for a drug developer of an extended release form to re-conduct safety and efficacy studies of the molecule. As such, the extended release formulation developer may rely on the safety and efficacy studies previously conducted and this developer need only show some clinical and non-clinical information to support the ER formulation.
505(b)(2) Application Approvals and FDA Exclusivities
Even though the (b)(2) application is a hybridized application, the sponsor may still earn FDA exclusivity. Typically, because the sponsor is not inventing any new molecule but repurposing an existing molecule, the FDA may award 3 year marketing exclusivity (e.g, for new dosage form, new patient populations, new indications, new combinations, etc.) or 7-year Orphan Drug Exclusivity (ODE) if there is a new orphan drug designation and exclusivity applicable.
The so-called New Chemical Entity (NCE) 5-year data exclusivity may also apply. The (b)(2) application can be the basis of a NCE 5-year data exclusivity. While typically the 5-year NCE exclusivity is through the 505(b)(1) process, it need not be so. Indeed, in 2020 about 18% of the (b)(2) approvals were for new molecular entities. The 505(b)(2) pathway may be useful when a drug product has been approved ex-USA, there’s a large body of scientific and safety literature about the drug product, but for some reason the drug was never US FDA approved. In these circumstances, the (b)(2) sponsor would rely on the ex-USA literature to help support the (b)(2) application and because this is a first time FDA approval, the sponsor may obtain 5-year NCE data exclusivity.
Review Timing of the 505(b)(2) Application
Because the (b)(2) application is a branded drug application versus a generic drug application, the sponsor will pay branded drug PDUFA user fees versus paying generic drug GDUFA user fees. The review cycle timing is, therefore, about 11 months. The Median Review Time since 2017 to 2020 have been 10, 13, 10, and 11 months respectively.
Intellectual Property Issues for the (b)(2) Application
The (b)(2) pathway can be used to create a new franchise of drug products. And when done properly, the FDA pathway will map alongside the IP and commercial strategy. Why does mapping matter? If the (b)(2) application is entitled to any FDA market exclusivity, then the question for the company or its investors is whether any additional protection can exist beyond the FDA market exclusivity. As such, it is important to know if any IP or commercial protection can exist beyond any 3/5/7 year market exclusivity.
To this end, it is important to build the right IP strategy to protect the sponsor’s investment. It would be unfortunate that any relevant IP is so old that it expires before any FDA exclusivity expires. If that happens, competitors could launch competing products right at the exclusivity expiration. It goes without saying that whatever the purpose of the (b)(2) application is that the purpose is covered by relevant IP. Sponsors need to work with skilled patent counsel on how to develop the “right” kind of IP during the development process to ensure that relevant IP is issued on time.
The Paragraph IV Process for 505(b)(2) Applications
Because the 505(b)(2) application will rely on an Reference Standard (RS)/Reference Listed Drug (RLD), the sponsor may need to certify to one or more Orange Book listed patents. As such, it is common that the Paragraph IV certification may be necessary. When a sponsor undertakes the Para. IV certification and notification process, there may be a subsequent lawsuit. Interestingly, the statute provides for a 30-month litigation stay. But with the PDUFA goal date of about 11 months, it is incumbent on the (b)(2) applicant to accelerate its litigation. Otherwise, it may sit on Tentative Approval for the next 19 months. Given that the (b)(2) drug product is a branded drug, this is all lost commercialization opportunity. Again, engaging skilled patent counsel during the development will help the sponsor address any Paragraph IV patents and strategize to accelerate any litigation.
The 505(b)(2) vs. ANDA
The easier application process is an obvious benefit of gaining approval through the 505(b)(2) pathway rather than the NDA pathway, but why is it better than the ANDA process for generics? Aside from any 3, 5, or 7 years of market exclusivity that is available for (b)(2) sponsors, the ANDA sponsor may obtain 180-days of market exclusivity. And even then the 180-day exclusivity for ANDA sponsors is more mythical than real given that rarely does a single ANDA filer enjoy sole 180-day exclusivity. Unlike the ANDA sponsor that is a so-called First Applicant, there is no automatic parallel in the (b)(2) realm.
Also, in certain drug products, an ANDA must copy the branded reference product (RLD) to be Q1/Q2 compliant. As such, the copying of the branded product may result in adverse formulation patent consequences. So, the 505(b)(2) pathway may allow certain ingredient switching to occur that may result in the company avoiding another’s patent and/or permits the acquisition of its own patents.
The approval timelines for the (b)(2) and ANDA are different, despite both paying user fees. The (b)(2) and the ANDA may be subject to litigation 30-month stays. The (b)(2) application also has the advantage that the sponsor need not copy the RLD’s label and be nearly identical. The ANDA label, however, is usually nearly identical to the RLD label. This means, strategically, that the (b)(2) applicant can draft a label to avoid either patents or other FDA exclusivities.
How we can help you?
We help 505(b)(2) applicants strategize the timelines for FDA approval, FDA market exclusivities, how the patent portfolio maps onto the product and timelines, how to expedite any patent litigation to resolution so that FDA approvals can be leveraged, and how arguments raised in any Paragraph IV litigation against any original brand company could backfire when the (b)(2) applicant is a plaintiff-patentee against later ANDA filers.
We help clients in patent litigation, appeals, counseling, opinions of counsel, and PTAB proceedings. When your current firm needs help or the client needs a change of counsel, we can help.
About Upadhye Tang LLP
Upadhye Tang LLP is an IP and FDA boutique firm concentrating on the pharmaceutical, life sciences, and medical device spaces. We help clients with navigating the legal landscape by helping on counseling and litigation. Clients call us to help move drug and device approvals along and to represent them in IP and commercial litigation. Call Shashank Upadhye, 312-327-3326, or by email: shashank@ipfdalaw.com, for more information.
No Attorney-Client Relationship or Legal Advice
The content of this website has been prepared by Upadhye Tang LLP for informational purposes only and does not constitute legal advice. Nothing shall not be construed as an offer to represent you, nor is it intended to create, nor shall the receipt of such information constitute, an attorney-client relationship. Please call us first with any questions about the firm.
Nobody should not take any action or choose not to take any action based upon the information on this website without first seeking appropriate professional counsel from an attorney licensed in the user’s jurisdiction. Because the content of any unsolicited Internet email sent to Upadhye Tang LLP at any of the email addresses set forth in this website will not create an attorney-client relationship and will not be treated as confidential, you should not send us information until you speak to one of our lawyers and get authorization to send that information to us.
Individual Opinions
The opinions and views expressed on or through this website, or any newsletter, are the opinions of the designated authors only, and do not reflect the opinions or views of any of their clients or law firms or the opinions or views of any other individual. And nothing said in any newsletter or website reflects any binding admission or authority.
